Hipertrofik Kardiyomyopatiler
Hipertrofik Kardiyomyopatiler : Hipertrofik Kardiyomiyopati (HKM), genellikle ailesel olan primer bir kalp hastalığıdır. Genel popülasyondaki prevalansı %0.2'dir. HKM, hipertansiyon veya kapak hastalığı gibi hipertrofi yapabilecek başka nedenlerin bulunmadığı, genellikle sol ventrikülde ve interventriküler septumda yerleşen asimetrik bir hipertrofiyle karakterize otozomal dominant bir hastalıktır. b-miyozin ağır zinciri, troponin T, troponin I, a-tropomiyozin, esansiyel ve regülatör hafif zincirler, kardiyak miyozin bağlayıcı protein C, aktin ve titin gibi sarkomerik proteinleri kodlayan genlerdeki mutasyonlar HKM'ye neden olurlar. Hastalık kardiyak yapıda ve klinik bulgularda değişikliklerle beliren çok çeşitli klinik görünümlere sahiptir. En ağır bulgusu, ani kalp ölümüdür. Bu çalışmanın amacı, kötü prognoz ve yüksek ani kalp ölümü insidansı gösteren b-miyozin ağır zincir genindeki Arg403Gln, Arg453Cys, Arg719Trp ve Arg719Gln mutasyonlarını saptamaktır. Çalışmaya klinik ve ekokardiyografik verileriyle HKM tanısı konulmuş 18 olgu alındı (9 erkek, 9 kadın, ort. yaş:39). Hastalardan alınan kanlardan fenol-kloroform ekstraksiyonu ve etanol çöktürme yöntemiyle DNA'lar elde edildi. Mutasyonların belirlenmesinde, PCR ve kısıtlayıcı enzim kesimi + mini horizontal agaroz elektroforez teknikleri kullanıldı. Çalışmada uluslararası literatürde en sık gözlenen mutasyonlar öncelikli olarak tarandı ve dünyada kabul edilen moleküler genetik yöntemler uygulandı. İncelenen 18 hastada Arg403Gln, Arg453Cys, Arg719Trp ve Arg719Gln mutasyonlarının olmadığı saptandı. Daha fazla sayıda hasta ve daha geniş imkanlarla HKM'den sorumlu olan mutasyonların belirlenerek bir ülke veri tabanı oluşturulmasıyla, preklinik tanı ve ani kalp ölümü için risk tayini yapılabileceği sonucuna ulaşıldı.
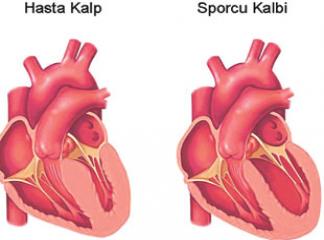
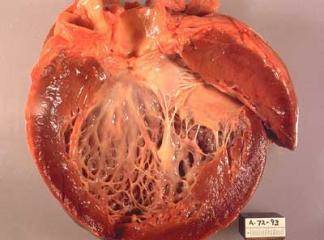
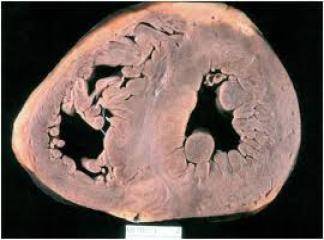
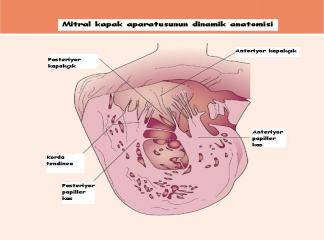
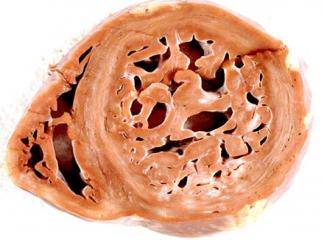
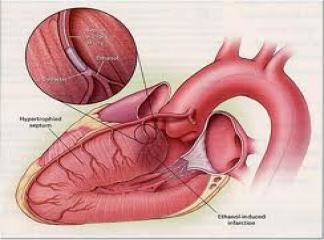
Hipertrofik kardiyomiyopati (HKM), en sık görülen kalıtımsal kardiyovasküler hastalıklardan biridir. Klinik, morfolojik ve genetik açıdan heterojenite gösteren HKM'nin en belirgin özelliği sol ventrikül hipertrofisidir. Hipertrofi, genellikle asimetrik ve interventriküler septum (İVS) yerleşimlidir. Ancak, kesin tanı için sol ventrikül hipertrofisine yol açabilecek başka bir patoloji (hipertansiyon, kapak hastalığı, koroner kalp hastalığı, aort koarktasyonu, konjenital kalp hastalığı) bulunmamalıdır (1).
HKM, %55 ailesel (Familyal Hipertrofik Kardiyomiyopati-FHK) formda olup, otozomal dominant kalıtım gösterir. Bunun dışındaki olgular sporadik olarak belirlenmektedir (2). Hastalığın klinik görünümü, asemptomatik durumdan, şiddetli kalp yetersizliği, hatta ani kalp ölümüne kadar uzanan geniş bir spektrum çizer (3). Özellikle sporcularda olmak üzere 35 yaşın altındaki gençlerde görülen ani kalp ölümünün en sık nedenidir (4). HKM'deki başlıca patolojik bulgu, sarkomer yapısında bozulma ve miyozit hipertrofisidir (5-7).
Hastalığın farklı toplumlarda ortalama görülme sıklığı %0.2'dir (4,8). Moleküler genetik tekniklerle HKM'ye yol açtığı saptanan 9 gen, b-miyozin ağır zinciri-[b-MAZ], troponin T, troponin I, a-tropomiyozin, regülatör ve esansiyel hafif zincirler, miyozin bağlayan protein C, aktin ve titin kalp kasının sarkomerinde bulunan farklı komponentleri kodlar (9). Bu genlerden biri olan b-MAZ genindeki mutasyonlara bağlı değişiklikler, FHK'nin %35'inden sorumludur (10).
b-MAZ genindeki değişik mutasyonlara sahip bireylerde hastalığın klinik görünümü ve şiddeti farklı olmaktadır. Arg403Gln, Arg453Cys, Arg719Trp ve Arg719Gln mutasyonlarına sahip bireylerde kötü prognoz, yüksek penetrans, yüksek ani kalp ölümü insidansı ve şiddetli sol ventrikül hipertrofisi gözlenir. Glu930Lys ile Arg249Gln mutasyonları ise ani kalp ölümü insidansı yönünden orta derecede bir risk taşırken, Leu908Val, Gly256Glu, Val606Met, Phe503Cys ve Arg723Cys mutasyonları benign bir prognoza sahiptirler (2,11-13).
Çalışmamızda, hastalığın moleküler temelini belirlemek üzere 18 HKM hastasında, b-MAZ genindeki Arg403Gln, Arg453Cys, Arg719Trp ve Arg719Gln mutasyonları incelendi.
Hipertrofik kardiyomiyopati (HKM), en sık görülen kalıtımsal kardiyovasküler hastalıklardan biridir. Klinik, morfolojik ve genetik açıdan heterojenite gösteren HKM'nin en belirgin özelliği sol ventrikül hipertrofisidir. Hipertrofi, genellikle asimetrik ve interventriküler septum (İVS) yerleşimlidir. Ancak, kesin tanı için sol ventrikül hipertrofisine yol açabilecek başka bir patoloji (hipertansiyon, kapak hastalığı, koroner kalp hastalığı, aort koarktasyonu, konjenital kalp hastalığı) bulunmamalıdır (1).
HKM, %55 ailesel (Familyal Hipertrofik Kardiyomiyopati-FHK) formda olup, otozomal dominant kalıtım gösterir. Bunun dışındaki olgular sporadik olarak belirlenmektedir (2). Hastalığın klinik görünümü, asemptomatik durumdan, şiddetli kalp yetersizliği, hatta ani kalp ölümüne kadar uzanan geniş bir spektrum çizer (3). Özellikle sporcularda olmak üzere 35 yaşın altındaki gençlerde görülen ani kalp ölümünün en sık nedenidir (4). HKM'deki başlıca patolojik bulgu, sarkomer yapısında bozulma ve miyozit hipertrofisidir (5-7).
Hastalığın farklı toplumlarda ortalama görülme sıklığı %0.2'dir (4,8). Moleküler genetik tekniklerle HKM'ye yol açtığı saptanan 9 gen, b-miyozin ağır zinciri-[b-MAZ], troponin T, troponin I, a-tropomiyozin, regülatör ve esansiyel hafif zincirler, miyozin bağlayan protein C, aktin ve titin kalp kasının sarkomerinde bulunan farklı komponentleri kodlar (9). Bu genlerden biri olan b-MAZ genindeki mutasyonlara bağlı değişiklikler, FHK'nin %35'inden sorumludur (10).
b-MAZ genindeki değişik mutasyonlara sahip bireylerde hastalığın klinik görünümü ve şiddeti farklı olmaktadır. Arg403Gln, Arg453Cys, Arg719Trp ve Arg719Gln mutasyonlarına sahip bireylerde kötü prognoz, yüksek penetrans, yüksek ani kalp ölümü insidansı ve şiddetli sol ventrikül hipertrofisi gözlenir. Glu930Lys ile Arg249Gln mutasyonları ise ani kalp ölümü insidansı yönünden orta derecede bir risk taşırken, Leu908Val, Gly256Glu, Val606Met, Phe503Cys ve Arg723Cys mutasyonları benign bir prognoza sahiptirler (2,11-13).
Çalışmamızda, hastalığın moleküler temelini belirlemek üzere 18 HKM hastasında, b-MAZ genindeki Arg403Gln, Arg453Cys, Arg719Trp ve Arg719Gln mutasyonları incelendi.
İLGİNİZİ ÇEKEBİLİR