Myelodisplastik Sendromlar
Bu sendroma sahip hastalarda, kemik iliği kısmen ya da tamamen mutasyona uğramış multipotent bir kök hücrenin klonal artışından kaynaklanan hücreler ile doludur. Bu kök hücre, eritrositlere, granulositlere ve trombositlere diferansiye olma kapasitesini korumakla birlikte, bu etkisiz ve düzensizdir. Sonuç olarak, kemik iliği genellikle hipersellüler ya da normosellülerdir, fakat periferik kanda bir ya da birden fazla hücre tipinin sitopenisi görülür. Kemik iliğindeki anormal kök hücre klonu genetik olarak dengesizdir ve ek mutasyonların birikimine açıktır. Bu nedenle AML'ye transformasyon olabilir. Vakaların çoğu idiopatiktir, fakat bazı hastalarda sendrom, alkilleyici ajanların kullanıldığı bir kemoterapiden ya da iyonize radyasyon tedavisinden sonra gelişir.
Sitogenetik çalışmalarda, hastaların yaklaşık %70'inde kemik iliğinde krornozomal olarak anormal bir hücre klonunun mevcut olduğu saptanmıştır. Sık görülen karyotipik anomaliler arasında, 5. ve 7. krornozomlann kaybı ve Sq ya da 7q'da delesyon sayılabilir. Morfolojik olarak, kemik iliği anormal görünümlü hematopoietik hücre öncülleri ile dolmuştur. Bazı sık görülen anomaliler arasında, Megaloblastik anemilerdekilere benzeyen megaloblastoid Eritroid öncüller, mitokondrilerinde demir birikimi içeren eritroid seri hücreleri (ring sideroblastlar), anormal granüller ya da nükleer matürasyon gösteren granülosit öncülleri ve tek küçük nükleolusa sahip küçük megakaryositler sayılabilir.
Hastaların büyük bir kısmı 50-70 yaş arası kişilerdir. Bunların % 10-40'ında AML gelişirken, geri kalanınında diferansiye myeloid hücre eksikliği nedeniyle devamlı olarak infeksiyonlar, anemi ve kanama görülür. Kemoterapiye cevap genellikle yetersizdir. Bu nedenle, myelodisplazinin bir kök hücre yetmezliği zemininden köken aldığı düşünülmektedir. Ayrıca, apıastik anemili bazı hastalarda sonunda myelodisplastik sendrom gelişmektedir. Oldukça az sayıdaki myelodisplazi hastasında da T hücre baskılayıcı ajanlara cevap görülmüştür. Bu ilişkiler, en azından hastaların bir kısmında, kemik iliğinde, normal kök hücreler T hücreleri tarafından saldınya uğradığından mutant bir klonun büyüyüp geliştiğini düşündürmektedir. Daha önce de bahsedildiği gibi, paroksismal noktümal hemoglobinüride de benzer bir mekanizma söz konusudur. Prognoz oldukça değişkendir. Ortalama yaşam süresi 9-29 ay arasında değişmektedir. Tanı konduğu sırada kemik iliğinde yüksek blast miktarı ve sitogenetik anomali varlığında prognoz daha kötüdür.
Kronik Myeloproliferatif Hastalıklar
Bu hastalıklarda, terminal diferansiasyon kapasitesine sahip neoplastik kemik iliği öncüllerinin hiperproliferasyonu söz konusudur. Sonuç olarak, periferik kanda bir ya da birden fazla tip matür elemanın artışı olur. Neoplastik öncüller sekonder hematopoietik organlara (dalak, karaciğer, lenf gangliyondan) da yerleşirler bunun sonucu olarak, hepatosplenomegali (neoplastik ekstra medüller hematopoiez nedeniyle), hafif lenfadenopati görülür. Bu hastalıklarda görülen ortak tema, normal myeloid hücrelerin büyümesini ve hayatta kalımını düzenleyen sinyalleri taklit eden sinyaller üreten mutant tirizin kinaz varlığıdır. Bu bulgu, myeloid hücrelerin fazla artışının nedeni açısından tatmin edici bir açıklama sağladığı gibi, tirozin kinaz inhibitörleri ile tedavi açısından da önem taşımaktadır.
Hastaların büyük bir kısmı, dört diagnostik antiteden birine dahildir: kronik myelojenik lösemi (KML), polisitemi vera (PCV), primer myelofibrozis ve esansiyel trombositemi. KML, karakteristik bir sitogenetik anomali olan BCR-ABL füzyon geni varlığı ile ilişkili olduğundan değerlerinden belirgin şekilde ayrılır. Diğer myeloproliferatif hastalıklar arasında ise klinik ve genetik açıdan önemli ölçüde örtüşmeler vardır. JAK2 kinaz mutasyonları bu grupta en sık görülen ortak genetik anomalidir. Bu anomali, polisitemi vera vakalarının %90'ından fazlasında, primer myelofibrozisti hastaların %SO'sinde ve esansiyel trombositemilerin %30'unda görülür. Myeloproliferatif hastalıkların daha ender görülen tipleri, platelet kaynaklı büyüme faktörü (platelet derived growth factor) alfa ve beta gibi diğer tirozin kinazlardaki aktive edici mutasyonlar ile ilişkilidir. Sonuç olarak, myeloproliferatif hastalıkların hepsinin olmasa da çoğunun, normalde hematopoietik büyüme faktörleri tarafından uyarılan bir tip tirerin kina: aktivitesinde anormal artış varlığı ile ilişkili olduğu söylenebilir. Burada sadece KML, PVC ve primer myelofibrozis anlatılacaktır. Esansiyel trombositopeni çok nadir görüldüğünden bu hastalıktan bahsedilmeyecektir.
Kronik Myelojenik Lösemi
KML, 2S ile 60 yaş arası yetişkinleri etkiler ve tüm lösemi vakalarının % IS-20'si oluşturur. En yüksek insidans hayatın dördüncü ve beşinci dekadlarındadır.
Patofizyoloji
Bütün KML vakalarında belli bir kromozomal anomali, BCR-ABLjüzyon geni mevcuttur. Vakaların %9S'inde BCR-ABL füzyon geni, kromozom 9'daki ABL genini kromozom 22'deki BCR geninin yakınlarındaki bir pozisyona taşıyan (9;22) translokasyonunun bir ürünüdür. Ortaya çıkan kromozom 22, ilk kez Philadelphia'da tanımlandığından Philadelphia (Ph) kromozomu olarak adlandırılır. Geri kalan %5 hastada BCR-ABL füzyon geni ikiden fazla kromozomun katılımı nedeniyle şifreli ya da bilinmeyen yeniden düzenlemeler sonucu ortaya çıkar. KML'li hastalarda, BCR-ABL füzyon geni, granülositik, eritroid, megakaryositik ve B hücre öncüllerinde, bazı vakalarda da
T hücre öncüllerinde mevcuttur. Bu bulgu KML'nin kaynak hücresinin pluripotent kök hücre olduğunun sağlam bir kanıtıdır. Bölüm 6'dan da hatırlanacağı gibi, BCR-ABL geni, BCR'nin ve ABR'nin tirazin kinaz alanının bölümlerini taşıyan ve neoplastik transformasyonda kritik rolü olan bir füzyon proteinini kodlar. Ph kromozomu KML için karakteristik olmakla birlikte, yetişkinlerdeki ALL'lerin %2S'inde ve az sayıda AML hastasında pozitif olduğu unutulmamalıdır.
Normal myeloid öncüller büyümeleri ve hayatta kalmaları için büyüme faktörleri ve bunların reseptörlerine bağımlıdırlar. Abncak KML öncüllerinin bu gereksinimleri daha azdır. Büyüme faktörü bağımlılığındaki bu azalma BCR-ABL tirozin kinazın varlığı nedeniyledir. BCR-ABL tirazin kinaz, büyüme faktörü reseptörü aktivasyonunun etkilerini taklit eden bir dizi sinyal üretir. BCR-ABL füzyon geni birden fazla hücre serisinde bulunsa da, henüz bilinmeyen nedenlerle, granülositik seri öncülleri en fazla etkilenen hücrelerdir. Kemik iliğinde ve periferik kanda granülositlerin belirgin şekilde artmasından anlaşılacağı gibi, prolifere olan KML öncülleri terminal diferansiasyon kapasitelerini korumaktadırlar.
Morfoloji
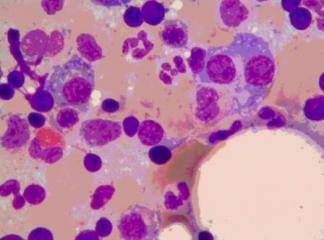
Periferik Kan bulguları karakteristiktir. Lenfosit sayısı artmış, sıklıkla 100.000 hücre/p], seviyesini geçmiştir. Dolaşımdaki hücreler baskın olarak nötrofiller, metamyelositler ve myelositlerdir (Şekil 12-26), fakat bazofiller ve eozinofiller de belirgin olabilir. Az miktarda myeloblast da genellikle %S'ten az oranda periferik kanda bulunabilir. Trombosit sayısında artış (trombositoz) da tipiktir. Granülositik ve megakaryositik seri öncüllerinde hiperplazi nedeniyle kemik iliği hipersellülerdir, Myeloblastlar genellikle hafif derecede artmıştır ve sıklıkla fagosit sayısında artış vardır. Büyümüş olan dalakta kırmızı pulca şiddetli ekstra medüller hematopoiez nedeniyle kemik iliğine benzemiştir. Hematopoietik hücrelerin oluşturduğu yer kaplayan kitle, lokal kan dolaşımını bozduğundan dalak enfartlarına yol açabilir.
Klinik Özellikler
KML genellikle yavaş başlangıçlıdır. İlk semptomlar nonspesifiktir (örn, çabuk yorulma, halsizlik, kilo kaybı). Bazen ilk semptom, ileri dereced splenomegaliden kaynaklanan kanrıda rahatsızlık hissidir. Bazen KML'yi, infeksiyon, stres, kronik inflamasyon ve bazı neoplazilerde meydana gelen ve belirgin granulosit artışı ile karakterize olan "lökomoid reaksiyon"dan ayırd etmek gerekir. KML ile lökomoid reaksiyon (ve diğer kronik myeloproliferatif hastalıklar) arasındaki ayrımda en önemli yardımcı, Ph kromozomu varlığıdır. Lökosit alkalin fosfataz seviyesi ölçümü de yararlı olabilir.Çünkü, KML'deki granülositler hemen hiçbir zaman alkalen fosfataz içermezken, lökomoid reaksiyonlarda ve diğer myeloproliferatif hastalıklarda (PVC gibi) bu enzim artmıştır.
KML yavaş ilerleyen bir hastalıktır. Tedavi verilmese bile ortalama yaşam süresi 3 yıldır. Belli bir süre sonra (bu sürenin ne kadar olacağı tahmin edilememektedir), hastaların yaklaşık % 50'si hızlı gidişli bir döneme girer. Bu dönemde, dereceli olarak artan bir tedaviye cevapsızlık söz konusudur, anemi ve trombositopeni artar, ek sitogenetik anomaliler ortaya çıkar ve sonunda akut lôsemiye benzer bir tabloya doğru bir değişim meydana gelir (blast krizi). Geri kalan %50 hastada, blast krizi, intermedier hızlanmış dönem olmadan aniden ortaya çıkar. Hastaların %30'unda, blast krizindeki hücreler pre-B hücreleridir. Bu bulgu ile, KML'nin pluripotent kök hücre kaynaklı olduğu bir kez daha desteklenmektedir. Geri kalan %70 hastada, blast krizi AML'ye benzer. Daha nadir olarak, KML, diğer myeloproliferatif hastalıklarda, özellikle de myelofibrozisli myeloid metaplazide görülene benzer yaygın bir kemik iliği fibrozisi dönemine ilerler.
KML tedavisi hızla gelişmektedir. Önceleri çoğu hasta, "hafif' bir kemoterapi ile palyatif olarak tedavi edilmekteydi. Bu tedavi, ne yazık ki, blast krizi gelişmesini engelleyemiyordu. Hastaların %70'inde iyileşmeyi sağlaması nedeniyle, kemik iliği transplantasyonu hala kesin tedavi yöntemidir. Ancak, uyumlu vericisi olmayan ve yaşlı hastalarda yüksek bir ölüm riski taşımaktadır. Bir BCRABL tirazin kin az inhibitörü olan Gleevec (imantirıib mesylate) stabil dönemdeki KML hastalarının büyük bir bölümünde tam remisyon sağlar ve Nonspesifik kemoterapi ajanlanna göre çok az toksiktir.
İmantinib mesylate kullanırken relapsa giren KML hastalannda, sıklıkla BCRABL'nin aktif bölgesinde ilacın bağlanmasına engelleyen yeni mutasyonlar meydana gelmiştir. Bu olay ilacın "hedefe yönelik" etki gösterdiğini kanıtlamaktadır. Bu ilacın tam kür sağlayıp sağlamadığını anlamak için daha fazla çalışma gereklidir. Ancak, kemik iliği transplantasyonu yapılamayan hastalarda mükemmel bir tedavi seçeneği olan bu ilaç hedefe yönelik kanser tedavilerinin gelişiminde büyük ilgi uyandırmıştır.
Polisitemi Vera
PCV'nin karakteristik özelliği, panmyelozise neden olan eritroid, granulositik ve megakaryositik elemanların neoplastik proliferasyonu ve matürasyonudur. Trombositler ve granülositler de artmış olmalarına rağmen, en belirgin klinik bulgular ve semptomlar eritrosit kitlesindeki mutlak artışa bağlıdır. Bu durum, hemokonsantrasyon sonucu oluşan rölatif polisitemiden ayırd edilmelidir. Bunun yanı sıra, mutlak polisiteminin reaktif formlarından farklı olarak, PVC‘de serumda eritropoietirı seviyesi düşüktür. Bu bulgu, neoplastik klonun eritropoietine ve diğer büyüme faktörlerine karşı olan hipersensitivitesinin bir yarı sımasıdır. Son yıllarda, hemen bütün vakalarda, PCV hücrelerinin, eritropoietin reseptörleri ve diğer büyüme faktörü reseptörlerinin sinyal yolağında rol oynayan bir tirozin kinaz olan JAK2'de özel bir mutasyon taşıdığı saptanmıştır. Bu mutasyon, 617. sırada valirı yerine fenilaIaninin yerleşmesine neden olur. PCV patogenezinde önemli rolü olan bu değişim, hücrelerin eritropoietik reseptörünün eritropoietine karşı hipersensitif hale gelmesi için yeterlidir.
Morfoloji
PeV'de başlıca anatomik değişiklikler, eritrositoz nedeniyle kan hacminin ve viskositesinin artışından kaynaklanır. Tüm dokularda ve organlarda pletorik konjesyon polisitemi vera için karakteristiktir. Karaciğer büyür ve sıklıkla ekstra medüller hematopoiez odakları içerir. Dalak, hastaların yaklaşık %75'inde hafif bir büyüme gösterir ve ekstamedüller hematopoiez ve vasküler konjesyon nedeniyle ağırlığı 250-300 gram'a kadar artar. Artmış viskositenin ve vasküler stazın sonucu olarak sıklıkla trombozlar ve enfarktüsler meydana gelir. Bunlar en sık olarak kalp, dalak ve böbrekleri etkiler. Bu hastaların yaklaşık üçte birinde, büyük olasılıkla kan damarlarının fazla genişlemesi ve anormal trombosit fonksiyonları nedeniyle kanamalar görülür. Bunlar genellikle gastrointestinal yol, orofarenks ve beyinde meydana gelirler. Her ne kadar bu kanamalar arada bir spontan olarak meydana gelse de, çoğunlukla minör travma ya da cerrahi bir müdahele sonrası ortaya çıkarlar. Neoplastik klon tarafından üretilen trombositlerde fonksiyon bozukluğu olabilir.
Trombosit defektinin yapısına göre, tromboza eğilim artar ya da anormal kanamalar görülür. KML'de olduğu gibi, periferik kanda bazofil sayısı artmıştır. Kemik iliği, eritroid, myeloid ve megakaryositik serilerin hiperplazisi nedeniyle hipersellülerdir, Buna ek olarak, hastaların %10'unda tanı konduğu sırada bir miktar kemik iliği fibrozisi de mevcuttur. Bazı hastalarda, hastalık myelofibrozise doğru ilerleme gösterir. Bu durumda kemik iliği boşluğu fibroblastlar ve kollajen dolduğu
Klinik Gidiş
Sinsi gidişli bir hastalık olan polisitemi vera genellikle geç orta yaşta görülür. Hastalar klasik olarak pletorik ve sıklıkla da siyanotiktir. Sayıca artmış olan bazofillerden histamin salınımı nedeniyle şiddetli pruritus görülebilir. Fazla histamin, bazı hastalarda görülen peptik ülserasyonun da sebebi olabilir. Diğer problemler tromboza ve hemorajiye eğilimden ve hipertansiyondan kaynaklanır. Baş ağrısı, baş dönmesi, gastrointestinal semptomlar, hematime: ve melena sıktır. Hücre döngüsünün hızlı olması nedeniyle, hastaların %5- 10'unda semptomatik gut görülür, fakat hastaların çok daha büyük bir kısmında semptomatik hiperürisemi vardır. Tam daha çok laboratuvar bulgulan ile koyulur. Eritrosit sayısı 6 ile 10 milyon/ul. arasında değişir, hematokrit ise %60'lara kadar ulaşabilir.
Kemik iliğinde granülositik öncüllerde ve megakaryositlerde de hiperproliferasyon olduğundan, granülosit sayısı 50.000/mm3'e kadar ulaşırken, trombosit sayısı sıklıkla 400.000/mm3'ten daha yüksektir. Bazofil sayısı da sıklıkla artmıştır. çoğu vakada trombositler fonksiyonel olarak anormaldir. Kanda dev trombositler ve megakaryosit parçaları görülebilir. Hastaların %30'unda, genellikle kalbi ve beyni etkileyen trombotik komplikasyonlar gelişir. Budd-Chiari sendromuna yol açan hepatik ven trombozu ender fakat ölümcül bir komplikasyondur. Minör kanamalar (örn, epistaksis ve diş eti kanamaları) oldukça sıktır, hayatı tehdit eden kanamalar ise hastaların %5- 10'unda görülür. Tedavi olmayan hastalar tanıdan sonra aylar içerisinde bu damarsal komplikasyonlar nedeniyle ölürler. Eğer eritrosit kitlesi, flebotomiler ile normale yakın seviyede tutulabilirse, ortalama yaşam süresi 10 yıla kadar uzatılabilir.
Hayatta kalım süresinin tedavi ile uzatılması sayesinde, PCV'nin doğal gidişinde, primer myelofibrozisin klinik ve anatomik özelliklerinin geliştiği bir "spent fazına (harcasına fazı) doğru dereceli bir transformasyon olduğu görülmüştür. Ortalama 10 yıllık bir dönemden sonra tümörlerin %15-20'sinde bu tür bir transformasyon görülür.Bu geçiş, kemik iliğinde ilerleyici fibrozis (myelofibrozis) ve hematopoiezin belirgin olarak büyümüş olan dalağa kayması ile karakterizedir. AML'ye benzer yapıda bir "blast krizi"ne transformasyon da KML'dekinden daha nadir olarak meydana gelebilir. Son yıllarda JAK2 inhibitörleri ile yapılan hedeflenmiş moleküler tedavi yöntemleri üzerinde durulmaktadır.
Primer Myelofibrozisli Myeloid Metaplazi
Bu kronik myeloproliferatif hastalıkta, periferik kanda beyaz hücre ve trombosit sayılarının yükseldiği kısa bir dönemden sonra, erken dönemde kemik iliği fibrozisi ile giden bir "spent fazı" gerçekleşir. Hematopoiez fibrotik kemik iliğinden, dalak, karaciğer ve lenf gangliyonlarına kayar. İleri derecede splenomegali ve hepatomegali gelişir. Bu ekstra medüller bölgelerdeki hematopoiez düzensiz ve etkisizdir. Kemik ili ği de fibrotik olduğundan, çoğu hastada orta derecede ya da şiddetli anemi ve trombositopeni görülür.
Bu durumun karakteristik özelliğinin kemik iliği fibrozisi olmasına rağmen, fibroblastların kaynağı transforme olmuş kök hücreleri değildir. Kemik ili ği fibrozisi hematopoetik hücrelerin, özellikle de megakaryositlerin düzeninin bozulmasına sekonder olarak meydana gelir. Kemik iliğindeki fibroblastların, neoplastik megakaryositlerden salgılanan platelet kaynaklı büyüme faktörü (platelet-derived growth factor) ve transforme edici büyüme faktörü f3 (transforming growth factor S) tarafından uyarılarak prolifere olduğuna inanılmaktadır. Bu iki büyüme faktörünün fibroblastlar için mitojenik olduğu bilinmektedir. Hasta klinik olarak semptom verdiğinde, fibroblastlar çoktan kemik iliğini kaplamış, ekstra medüller hematopoiez belirgin hale gelmiştir. Daha az sıklıkla, kemik iliği fibrozisi tanı sırasında belirgin değildir. Bu hastalarda, diğer "hiperproliferatif" myeloproliferatif hastalıkların tipik klinik tablosu görülür.
Primer myelofibrozis (ve esansiyel trombositemi) vakalarının yaklaşık yarısında, PVC‘de görülen JAK2 mutasyonunun (617. amino asit sırasında valin yerine fenilaıaninin yerleşimi mutasyonu) aynısının varlığının patojenik ve olasılıkla törapötik önemi vardır. Bu bulgu, bu antiteler arasında ne kadar geniş bir örtüşme olduğunu göstermektedir. Aynı mutasyonu gösteren tümörlerde neden farklı klinik tablolar ortaya çıktığı bilinmemektedir. Belki primer myelofibroziste JAK2 mutasyonu farklı bir kök hücre popülasyonunda meydana gelmektedir, ya da bazı kişilerde spent fazının erken dönemde ortaya çıkması sadece bir tesadüftür
Morfoloji
Primer myelofibrozisli myeloid metaplazide ekstra medüller hematopoiezin gerçekleştiği başlıca bölge olan dalak belirgin olarak büyümüştür. Ağırlığı bazen 4000 grama kadar ulaşabilir. Masif splenomegali nedeniyle multipl subkapsüler infarktlar bulunabilir. Histolojik olarak, dalakta normoblastlar, granülosit öncülleri ve megakaryositler mevcuttur ve bunlar sıklıkla anormal mortolojiye sahip sayıca artmış hücrelerdir. Bazen üç ana hücre serisinden birinin aktivitesinin orantısız olarak diğerlerinden farklı olduğu görülür. Orta derecede büyüme gösteren karaciğerde ekstra medüller hematopoiez odakları mevcuttur. Mikroskopik olarak, lenf gangliyonları da ekstra medüller hematopoiez odakları içerirler. Fakat bu, lenf gangliyonlarının genişlemelerine yetecek kadar fazla değildir.
Tipik bir vakada kemik iliği hiposellülerdir ve diffüz fibrozis gösterir. Ancak, erken dönemlerde kemik iliğinde her üç hücre serisinin de eşit olarak arttığı bir hipersellülerime izlenir. Sıklıkla belirgin olan megakaryositler displastik değişiklikler gösterebilirler.
Klinik Gidiş
Primer myelofibrozis, PCV ya da KML gibi bir kan tablosu ile başlayabilir, fakat daha sıklıkla, klinik olarak dikkati çekene kadar kemik iliğinde fibrozis gelişmiştir. çoğu hastada orta-ağır derecede anemi vardır. Beyaz kan hücresi sayısı normal olabileceği gibi, düşük ya da yüksek de olabilir. Hastalığın erken dönemlerinde trombosit sayısı normal ya da yüksektir, fakat sonunda bütün hastalarda trombositopeni gelişir. Periferik kan yayması belirgin şekilde anormaldir. Eritrositler ile ilgili anomaliler arasında, nükleus içeren garip şekilli eritrositlerin (poikilositler, gözyaşı hücreleri) varlığı başta gelir. Nükleuslu eritroid seri öncülleri de periferik kanda görülebilir. İmmatür beyaz kan hücreleri (myelositler ve metamyelositler) ve bazofiller genellikle artmıştır. Nükleuslu eritrosit öncüllerinin ve immatür beyaz kan hücrelerinin varlığı lökoeritrositozis olarak adlandırılır.
Trombositler sıklıkla anormal boyutlarda, anormal şekilli olup, fonksiyonları bozuktur. Bazı vakalarda klinik tablo ve kan bulguları KML'ninkilere benzer, fakat Ph kromatomu yoktur. Hücre döngüsünün çok hızlı olması nedeniyle ortaya çıkan hiperürisemi ve gut tabloyu tamamlayan diğer özelliklerdir. Myeloid metaplazinin sonuçları oldukça değişkendir. Ortalama yaşam süresi 4-5 yıldır. Enfeksiyon tehditi, ve trombosit anomalileri nedeniyle trombotik ve hemorajik ataklar mevcuttur. Dalak enfarktüsleri sıktır. Hastaların yaklaşık %5-15'i sonunda AML'ye benzeyen bir blast krizine girer.
İLGİNİZİ ÇEKEBİLİR