Lizozomal Depo Hastalıklar
Bilindiği gibi, lizozomlar fıngolipidler ve mukopolisakkaridIer gibi kompleks ub tratların çözünebilir on ürünlere parçalanmasında kullanılan değişik hidrolilik enzimleri içerirler. Bu substratlar ya hücre i organellerin dönüşümünden ortaya çıkıp otofago itozla lizozomların içine alınır, ya da hücre dışından fago itozla alınır. Lizozomal bir enzimde kalıt al bir eksiklik olduğunda. onun ub tratının katabolizması tamamlanamaz, bu deki men par alanmış çözünmeyen metabolitlerin lizozomların i inde birikmesine neden olur.Bugüne kadar yaklaşık 40 lizozomal depo hastalığı tanımlannuştır.
Bunların herbiri spesifık bir lizozomal enzimin veya proteinin fonksiyonel eksikliğiyle ortaya çıkmaktadır. Klasik olarak. lizozomal depo hastalıkları substratların ve biriken metabolitlerin biyokimyasal özelliklerine göre sınıflandırılmaktadır, ancak altta yatan moleküler bozukluğa bağlı bir sınıflama da yapılabilir.Her grupta herbiri pesifik bir enzim eksikliği sonucu ortaya çıkan birkaç hastalık vardır. Aslında karmaşık görünmesine rağmen, bu gruptaki birçok hastalıkta ortak bulgular vardır:
• Otozornal resesif kalıtım
• Sıklıkla bebek ve küçük çocukların etkilenmesi
• Çözünmeyen ara ürünlerin mononükleer fagositer sistemde depolanarak hepatosplenomegaliye neden olması
• öronal ha arla birlikte sık santral sinir sistemi tutulumu
• Hücresel disfonksiyon (bu hem sindirilemeyen materyalin birikimi, hem de makrofaj aktivasyonu ve sitokin salının gibi ikincil gelişen olaylar nedeniyledir)Bu hastalıkların çoğu oldukça nadirdir, detaylı tanımlamalarına çeşitli kitaplar ve derlemelerde yer verilmiştir.Burada sadece daha sık olan hastalıkların birkaç tanesinden bahsedilecektir. Aynı zamanda bir lizozomal depo hastalığı olan tip II glikojen depo hastalığı (Pompe hastalığı) daha sonra anlatılacaktır.
Tay-Sachs Hastalığı (GM2 Gangliosidoz: Heksozarninidaz a-Subünite Eksikliği)
Gangliosidozlar katabolık bir lizozomal enzim eksikliğinde ortaya çıkan ve özellikle beyinde gangliosidlerin birikimiyle karakterize ha talıklardır. Etkilenen ganglioside bağlı olarak bu hastalıklar Gmı ve GM2 olmak üzere iki alt gruba aynlır. Tüm ganglio idozların en sık görüleni olan Tay-Sachs hastalığı GM2'nin parçalanmasından sorumlu olan heksozarninidaz A enzimininn subünitesinin eksikliğiyle karakterizedir. Bugüne kadar 90'dan fazla mutasyon tanımlanmıştır ve bunların çoğu proteinlerin hücre içi taşınması veya katlanmasını etkilemektedir. Bu hastalıkta gangliosid metabolizmasının en sık görüldüğü organ olan beyin, başlıca etkilenen organdır.
Tüm santral sinir sisteminde nöronlar, sinirlerin akson silindirleri ve glial hücrelerde GM2 depolanması olur. Etkilenen hücreler ödemli ve köpüksü görünürler.Elektron mikroskopisinde lizozomlann içinde çevrimsel yapılar (whorl) saptanır.Bu anatomik değişiklikler medulla spinalis de dahilolmak üzere tüm santral sinir sistemi boyunca, periferik sinirlerde ve otonom sinir sisteminde görülür. Retina da genellikle etkilenmiştir.
Nöronal hasarın moleküler temeli bugün için tam olarak anlaşılamamıştır. Birçok vakada mutant protein katlanmasında bozukluk olduğu için, katlanmanuş protein cevabı (unfolded protein response) olarak adlandınlan bir mekanizma tetiklenir. Eğer katlanması bozuk proteinler şaperonlar tarafından stabilize edilemezse apoptozu kolaylaştırırlar. Bu bulgular, bu ve benzeri lizozomal depo hastalıklarında şaperon tedavisini bir seçenek olarak ortaya koymaktadır.
Diğer lipidozlar gibi, Tay-Sachs hastalığı da en sık Aşkenazi Yahudilerinde görülür. Bunlarda heterozigot taşı yıcıların sıklığı 30'da 1 olarak tahmin edilmektedir. Serum heksozaminidaz düzeyi ölçülerek veya DNA analizi ile heterozigotlar belirlenebilir. Tay-Sachs hastalığının en sık görülen akut infantil formunda bebekler doğumda noraldir, ancak 3-6 ay içinde motor güçsüzlük başlar, mental retardasyon, körlük ve ağır nörolojik bulgular eklenerek hastalar iki üç yıl içinde kaybedilirler.
Niemann-Pick Hastalığı, Tip A ve B
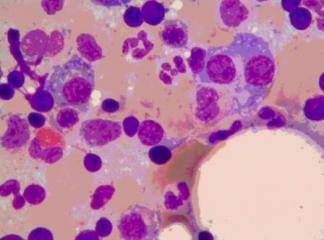
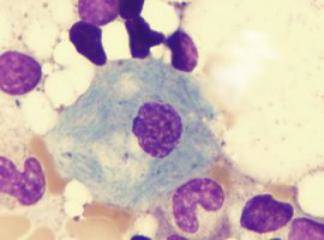
Birbiri ile ilişkili bu iki hastalık primer asit sfıngomyelinazın eksikliği ve bunun sonucunda gelişen sfingomyelin birikimi ile karakterizedir. Tip A, sfingomyelinazın ağır eksikliği ile karakterize olup, sfıngomyelirıin seramid ve fosforilkoline parçalanmasında bozukluk vardır. Bütün fagositik hücrelerde ve nöronlarda aşırı sfingomyelin birikimi olur. Makrofajlar bu kompleks lipid partikülleri ile dopdolu hale gelir, bu da sitoplazmalarına ince vakuoler veya köpüksü bir görünüm verir (Şekil 7-7). Çok sayıda fagositik hücre içeren dalak, karaciğer, kemik iliği, lenf nodları ve akciğerler en ağır etkilenen organlardır. Dalak büyümesi çok belirgin olabilir. ayrıca, medulla spinalis ve gangliyonlar da dahil olmak üzere tüm santral sinir sistemi de bu hastalıktan etkilerıir. Etkilenmiş nöronlar lipid depolanmasına bağlı olarak genişlemiş ve vakuollüdür, Hastalığın bu tİpi bebeklik döneminde masif organomegali ve ağır nörolojik bozulma ile kendini gösterir. Hastalar genellikle hayatın ilk üç yılı içinde kaybedilir.ü
Tip B' de ise organomegali vardır ancak nörolojik bulgular görülmez. Ha talıktan şüphelerıilen vakalarda ve taşıyıcıların aptanmasında lökositlerde veya fibroblast kültüründe sfıngomyelinaz aktivitesine bakılır. Antenatal taru enzim çalışma ı veya DNA analizi ile mümkündür.
Niemann-Pick Hastalığı, Tip C
Önceleri tİp A ve B ile ilişkili olduğu düşünülen bu hastalık aslında biyokimya al ve moleküler olarak farklıdır ve diğerlerinden daha sık görülmektedir. NPCl ve NPC2 genlerindeki mutasyonlarla ortaya çıkar. Vakaların çoğunda NPCl sorumludur. Diğer birçok lizozomal depo hastalığının aksine, Niemarın-Pick tip C primer olarak lipid taşınmasındaki bir bozukluğa bağlı ortaya çıkmaktadır. Etkilenen hücrelerde kolesterolün yanısıra GM1 ve GM2 gibi gangliosidler de birikir.
NPCl genirıin etkilediği biyokimyasal basamak bugün için hala kesin olarak bilinmemektedir. Hastalık klinik olarak heterojendir. En sık görülen formu çocukluk çağında ortaya çıkar ve ataksi, vertikal yukarı bakış kısıtWığı,distoni, disartri ve psikomotor gerileme ile gider.
Gaueher Hastalığı
Glukozilserarnidazı kodlayan gendeki mutasyonlar sonucu ortaya çıkar. Gaucher hastalığırun farklı allelik mutasyonlar sonucu ortaya çıkan beş otozomal resesif formu vardır. Hepsinin ortak özelliği serarnidden glukozu ayıran glukozilserarnidaz enziminde değişik derecelerde eksiklik olmasıdır. Bu enzim eksikliği mononükleer fagositik hücrelerde glukozilserarnid birikimine,bu hücrelerin Gaucher hücresi olarak adlandırılan hücrelere dönüşmesine neden olur. Normalde eski kan hücreleri,özellikle de eritrositlerin parçalanmasıyla ortaya çıkan glikolipidler belli bir düzen içinde parçalanır.Gaucher hastalığında ise bu yıkım glukozilseramidler düzeyinde durur. Bunlar dolaşırında makromoleküller olarak taşırımaları sırasında özellikle karaciğer, dalak ve kemik iliğindeki fagositik hücreler tarafından alınırlar. Bu fagositler (Gaucher hücreleri) şişkinleşmiş lizozomların birikimi nedeniyle bazen 100 uru'ye kadar genişler ve "buruşrnuş kağıt"olarak karakterize edilen patognomonik bir sitoplazmik görünüm alır.
Bu hücrelerde belirgin vakuolizasyon yoktur. Bugün artık Gaucher hastalığının oluşmasında sadece biriken materyalin değil, aynı zamanda makrofaj aktivasyonunun da önemli olduğu bilinmektedir. Etkilenen dokularda makrofajlardan salınan interlökinler (IL-2, IL-6) ve tümör nekrozis faktör (TNF) gibi sitokinler yüksek oranda bulunmaktadır.
Kronik nöronopatik olmayan form olarak da adlandırılan tip I, Gaucher hastalığı vakalarının %99'unu oluşturmaktadır. Hastaların %70 ile 100'ünde klinik veya radyolojik kemik tutulumu (osteopeni, fokallitik lezyonlar, osteonekroz) ile karakterizedir. Diğer bulgusu hepatosplenomegalidir; santral sinir sistemi tutulumu olmaz. Dalak genellikle bütün karını dolduracak kadar büyümüştür. Karaciğer, dalak, lenf nodları ve kemik iliğinde Gaucher hücreleri mevcuttur. Kemik iliği tutulumu ve kortikal erozyon hem radyolojik olarak da saptanabilen kemik lezyonlarına,hem de kanın şekilli elemanlarında azalmaya neden olabilir. Kemik değişikliklerinden, yukarıda bahsedilen makrofajlardan salınan sitokinlerin sorumlu olduğu düşünülmektedir. Tip I sıklıkla Aşkenazi Yahudilerinde görülür ve diğer tiplerden farklı olarak bu hastaların yaşam süresi uzundur. Tip II ve tip III ise nörolojik belirti ve bulgularla karakterizedir.
Tip II'de semptomlar 2 yaşından önce başlar ve daha ağırdır. Tip III' te ise semptomlar daha geç ortaya çıkar ve daha hafiftir. Karaciğer ve dalak tutulumu da olmasına karşın, klinikte nörolojik bulgular hakimdir. Ayrıca hastalığın hepatosplenomegali, cilt buluları ve non immün hidrops ile karakterize perinatal-ölümcül formu vardır. Kardiyovasküler form adı verilen bir diğer formda ise mitral ve aort kapak tutulumu ve kalsifikasyonu görülür.
Hastalığın tanısında ve heterozigotların saptanmasında lökositlerde veya fibroblast kültüründe glukozilseramidaz düzeyinin ölçümü yararlıdır. Güncel tedavi olarak saflaş tırılrruş enzim infüzyonu ile enzim replasmanı amaçlanmaktadır. Yeni bir tedavi seçeneği ise glukozilseramid sentetazı baskılayan ilaçlarla glukozilseramid birikimini önlemektir. Böylece glukozilseramid yapımı, dolayısıylada birikimi azalacaktır. Daha ilerisi için ise normal gen ile transfekte edilmiş otolog hematopoetik kök hücre infüzyonuyla glukozilserarnidaz gen tedavisi gündemdedir.
Mukopolisakkaridozlar
Mukopolisakkaridozlar (MPS) mukopolisakkaridlerin parçalanmasında bozukluk ve dolayısıyla değişik dokularda mukopolisakkarid birikimi ile karakterize hastalıklardır. Mukopolisakkaridler bağ dokusu ara maddesinin bir kısmını oluşturmakta ve bağ dokusundaki fibroblastlar tarafından sentezlenmektedir. Bunların çoğu ara maddeye salgılanırken,.bir kısmı da Iizozomların içinde yıkılır. Bu katabolik yolda birkaç enzim yer alır.Bu enzimlerin eksikliği lizozomlarda mukopolisakkarid birikimine neden olur. Her biri spesifık bir enzim eksikliği sonucu ortaya çıkan mukopolisakkaridozların MPSI'den MPS VII'ye kadar numaralandırılarak sınıflanan klinik formları vardır. Dokularda biriken mukopolisakkaridler dermatan sülfat, heparan sülfat, keratan sülfat ve bazı vakalarda kondroitin sülfattır.
Genel olarak MPS'ler karaciğer, dalak, kalp ve kan damarları gibi organları da içeren, birden çok organ tutulumuyla karakterize, ilerleyici hastalıklardır. Çoğunda kaba yüz görünümü, korneada bulanıklaşma, eklem sertliği ve mental retardasyon vardır. Biriken mukopolisakkaridlerin idrarla atılımı genellikle artmıştır. X'e bağlı resesif olarak kalıtılan Hunter endromu dışında, diğer tüm mukopolisakkaridozlar otozomal resesif hastalıklardır. Hastalığın tanımlanmış olan yedi tipinden burada sadece iyi bilinen iki sendroma kısaca yer verilecektir.Mukopolisakkaridoz tip I hepsi a-L-iduronidaz eksikliğiyle ortaya çıkan ve klinik olarak hafiften ağıra kadar değişiklik gösteren üç hastalığı içerir. Bu spektrumun iki ucunda Hur1er ve Scheie sendromları, ortada i e HurlerScheie sendromu yeralır.
Hurler sendromunda etkilenen çocukların beklenen yaşam süresi 6-10 yıldır. MPS'lerin birçok diğer formunda olduğu gibi, hastalarda kaba yüz görünümü ve iskelet deformiteleri gelişir. Olüm genellikle kardiyak komplikasyonlarla olur, ki bunlar da koroner arterler ve kalp kapakçıklarında mukopo isakkarid birikimiyle oluşan kabarık endotelyal ve endokardiyallezyonlara bağlı gelişir, Mononükleer fagositer sistem hücreleri,fibroblastlar, damar duvarındaki düz kas hücreleri ve damar endotelinde dermatan sülfat ve heparan sülfat birikimi olur. Etkilenen hücreler şişkin ve berrak sitoplazmalıdır; bu görünüm vakuollü lizozomlarda period ikasitSchiff (PAS) pozitif materyal birikimi nedeniyle ortaya çıkmaktadır. Lizozomal inklüzyonlar nöronlarda da bulunur ve bunlar mental retardasyondan sorumludur.
Tip II veya Hunter sendromu olarak adlandırılan diğer bir MPS çeşidi ise Hurler sendromundan kalıtım şeklinin X'e bağlı olması, korneada bulanıklaşma olmaması ve genellikle daha hafif klinik seyri ile ayrılır. Hurler sendromunda olduğu gibi Hunter sendromunda da biriken mukopolisakkaridler heparan sülfat ve dermatan sülfattır, ancak burada L-idüronat sülfataz eksikliği mevcuttur. Enzim eksikliklerinin farklı olmasına rağmen aynı maddelerin birikmesi, dermatan sülfat ve heparan sülfatın yıkımında aL-idüronidaz ve sülfatazın ikisinin birden gerekli olmasırıdandır. Eğer bu enzimlerden biri eksikse daha ileri yıkım olamaz
İLGİNİZİ ÇEKEBİLİR