Lipid depo hastalıkları
A. Gaueher Hastalığı
Gaucher hastalığı en sık lipid depo hastalığıdır. GIikoserebrozidaz enzim üretimini otozomal resesif kalıtımsal eksikliğinin bir sonucudur. Bu enzim makrofajlardaki lizozimlerde glikolipid yıkımı için gereklidir. Bu enzimin eksiklik veya bozukluğunda çözünemeyen glikoserebrozit sitoplazmada birikir.
Gaucher hastalığı Avrupa'nın kuzey ve ortalarında yaşayan Askenazi Yahudilerinde en sık görülür. Nadiren İsveçlilerde de sık görülebilir. Askenazi Yahudilerinde homozigot görülme oranının 800'de 1 olduğu tahmin edilmektedir. En sık mutasyon enzimin daha az üretimi ile sonuçlanan ve kromozom 1'in kısa kolunda yerleşen mutasyondur. Hastalığın ağırlığını mutasyon durumu belirler ve infant döneminde tanı alacak kadar ağır seyirli olabildiği gibi erişkin yaşta taramalarda saptanan asemptomatik formda da olabilir.
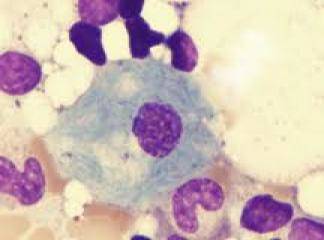
1. Klinik alttipleri; gaucher hastalığının 3 klinik alttipi tanımlanmıştır. Tip 1 en sık görüleni olup vakaların %99'una karşılık gelir. Aynı zamanda en heterojen grup olup hastalık dalak, ilik ve karaciğer makrofajlarında sınırlıdır. Tip 2 ise çok daha ağır bir formdur ve infant döneminde fulminan nörolojik tablo ile tanı alır ve hastalar genelde ilk 18 ay içinde ölür. Tip 3 çocukluk döneminde nörolojik bulgular ile tanı alır ama çok daha uzun bir yaşam beklentisi vardır.
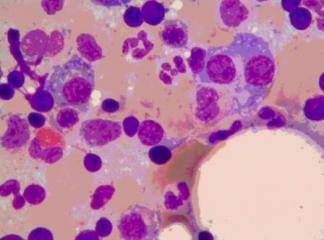
Tip 1 hastalar genelde hepatosplenomegali, kemik anormallikleri, anemi, pansitopeni, ilerleyici karaciğer disfonksiyonu ile gelirler. İlikte çok sayıda gaucher hücrelerinin görülmesi genelde tanı koydurucudur. Diğer önemli bulgular uzun kemiklerin uçlarında erlenmayer tipi deformite ve artmış serum asit fofataz aktivitesidir. Tanıda şüphe varsa periferik lökositlerde ß-glukosidaz ölçümü yapılabilir.
Gaueher benzeri hücreler yaşlılarda ve lenfoproliferatif veya myeloproliferatif hastalığı olanlarda da görülebilir. İstisna olarak gaucher benzeri hücreler AIDS hastalarının iliklerinde de görülebilir. Bu ß-glukosidaz aktivitesinin azalmasından değil makrofajların çok fazla hücre artığını alması ve bunun sitoplazmada birikmesinden kaynaklanır.
2. Prenatal ve genetik tanı; gaucher hastalığı genetik geçişli olduğundan amniotik sıvı hücrelerinde f3-g1ukosidaz aktivitesi ölçülerek veya genetik analizler ile prenatal tanı sağlanabilir. En sık görülen mutasyon ile ilgili olarak bir polimorfizm uzunluk sınırlaması vardır. Gaucher hastalığı için heterozigot olanların yarısında enzim normal olup kesin tanı genetik analizler ile yapılabilir. Bunların iliğinde gaucher hücresi yoktur ve klinik olarak normaldirler.
B. Niemann-Pick ve Tay-Sachs hastalığı
Lipid depo hastalığının diğer nadir formları olup yine Askenazi Yahudilerinde sık görülme eğilimindedir. Bu da glikolipid depo hastalığı için tanımlanan dengeli polimorfizm ile uyumludur. Nieman-pick' de makrofajlarda sfingomyelin birikimi ilik, lenf nodu ve diğer organlarda görülür. Tay-Sachs hastalığında ise merkezi sinir siteminde GM-2 gangliozit birikir. Bu iki durumda infant döneminde ağır nörolojik bulgular ile tanı alır. Etkin tedavisi olmadığı için genetik danışma, prenatal tanı ile önleme önerileri.
İLGİNİZİ ÇEKEBİLİR