Küçük Lenfositik Lenfoma Kronik Lenfositik Lösemi
Bunlar aslında aynı tümörlerdir, aralarındaki tek fark periferik kan tutulumu varlığıdır. Periferik kanda lenfositoz 4000 hücre/RNA-'u geçmiş ise kronik lenfositik lösemi (KLL) tanısı konur. Dolaşırnda büyük miktarda tümör hücresi yok ise küçük lenfositik lenfoma (SLL) olarak adlandınlırlar. Batı dünyasında yetişkinlerde en sık görülen lösemi ALL‘dir. SLL ise NHLlerin sadece %4'ünü oluşturmaktadır. Tam olarak açıklanamayan nedenlerle hem KLL hem de SLL Asya ülkelerinde ise oldukça nadirdir.
Patofizyoloji
Neoplastik B hücreleri henüz tam olarak anlaşılamarınş mekanizmalar üzerinden normal B hücre fonksiyonlanm baskılarlar. Bu nedenle, KLL/SLL'li hastaların çoğunda hipogamaglobulinemi görülür. Diğer taraftan, hastaların yaklaşık %l5'inde otolog eritrositlere karşı antikorlar mevcuttur. Diğer otoantikorlar da saptanabilir. Bu otoantikorlar tümöral olmayan B hücreleri tarafından üretilirler. Bu durum, bağışıklık sisteminin düzenlemesinde genel bir bozukluk olduğunu göstermektedir. Zaman geçtikçe tümör hücreleri normal kemik iliği elemanlanın yerini alırlar ve anemi, nötropeni ve trombositopeni meydana gelir. Gelişmesi nedeniyle hemolitik anemi ortaya çıkar.
Morfoloji
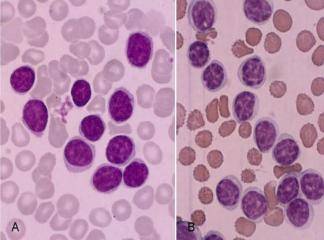
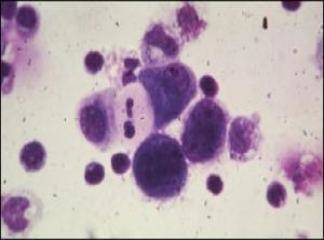
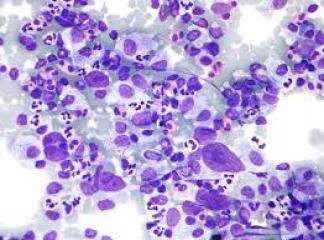
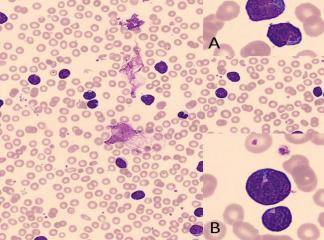
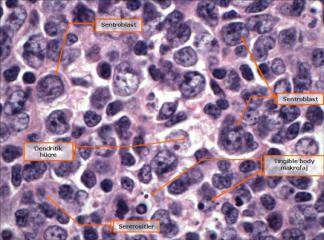
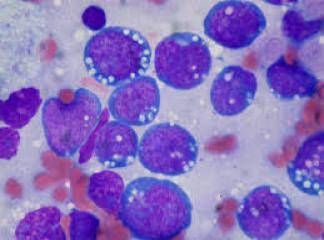
SLUKLL'de tutulan lenf gangliyonlarında yaygın olarak, küçük lenfositlerden oluşan tabakalar ve prolenfosit adı verilen büyük hücrelerin meydana getirdiği sınırları belirgin olmayan odaklar içermektedir. Baskın olan hücreler, koyu boyanan yuvarlak nukleuslara, dar sitoplâzmalara sahip olmaları ve boyut farklılıkları olmaması nedeniyle uyarı almamış görünümlü küçük hücrelerdir. Mitotik olarak aktif prolenfositlerden olu-şan odaklar proliferasyon merkezleri olarak adlandırılırlar. Bunların varlığı KLUSLL için patognomoniktir. Proliferasyon merkezleri dışında mitoz ender olarak görülür ve sitolojik atipi ya hiç yoktur ya da çok azdır. Hemen bütün vakalarda, lenf gangliyonlarına ek olarak, kemik iliği, dalak ve karaciğer tutulumu mevcuttur. Çoğu hastada, küçük, matür görünümlü lenfositlerden oluşan mutlak lenfositoz vardır. Neoplastik lenfositler kırılgan olduklarından sıklıkla yayma hazırlanışı sırasındaki mekanik etkiler nedeniyle parçalanırlar ve karakteristik is lekesi (smudge) hücrelerini oluştururlar. Daha büyük olan prolenfositler de kan yaymalarında görülebilirler
İmmunfenotip, Karyotip ve Moleküler Özellikler
KLL/SLL, pan-B hücre belirleyicileri olan CD19, CD20, CD23‘ü ve yüzey immunglobülinleri ağır ya da hafif zincirleri eksprese eden matür B hücrelerinin bir neoplazisidir. Tümör hücreleri CDS‘ de eksprese ederler. Bu özellik, B hücre neoplazileri arasında sadece mantle hücreli lenfoma ile ortaktır. Hastaların yaklaşık %SO'sinde karyotipik anomaliler görülür. Bunlardan en sık görülenleri trisomi 12 ve 11. ve 12. kromozomlardaki delesyonlardır. Diğer lenfoid neoplazilerden farklı olarak, kromozomal translokasyonlar nadirdir. Çoğu KLL/SLL'de immunglobülin segmentlerinde somatik hipermutasyon vardır. Bu bulgu, hücrelerin post-foolikül merkezi B hücresi (büyük olasılıkla hafıza hücresi) kaynaklı olduğunu göstermektedir. Daha nadir olarak, tümör hücreleri follikül merkez reaksiyonu göstermemiş olan naive B hücrelerinden kaynaklanırlar. Bu tümörlerin prognozu daha kötüdür.
Klinik Özellikler
KLL/SLL genellikle tam konulduğunda asemptomatiktir. Semptom görüldüğünde ise, sıklıkla nonspesifık olan bu semptomlar arasında kolay yorulma, kilo kaybı ve iştahsızlık sayılabilir. Jeneralize lenfadenopati ve hepatosplenomegali vakaların %SO-60'ında mevcuttur. Total lökosit sayısında hafif bir artış olabileceği gibi, bu sayı 200.000/ı-TL‘ye kadar artabilir. Hastaların yaklaşık %50'sinden fazlasında hastalığın geç dönemlerinde görülen hipogamaglobulinemi nedeniyle bakteriyel infeksiyonlara eğilim artmıştır. Daha nadiren, oto immun hemolitik anemi ve trombositopeni görülebilir. Hastalığın gidişi ve prognozu oldukça değişkendir. Çoğu hasta, tanının konulmasından sonra 10 yıldan fazla yaşar ve bu hastalık ile ilişkisi olmayan sebeplerden ölür. Ortalama hayatta kalma süresi 4-6 yıldır. Bununla birlikte, KLUSLL zamanla promyelositik lösemi ya da diffüz büyük B hücreli lenfomaya benzer daha saldırgan tümörlere dönüşmeye meyilli hale gelir. Transformasyon gerçekleştiğinde ortalama hayatta kalma süresi 1 yıldan azdır.
İLGİNİZİ ÇEKEBİLİR
Yorumlar
Ayn** Kal**********
6 yıl önce